|
THE REACTION BETWEEN METHANE AND CHLORINE
A Free Radical Substitution Reaction This page gives you the facts and a simple, uncluttered mechanism for the free radical substitution reaction between methane and chlorine. If you want the mechanism explained to you in detail, there is a link at the bottom of the page. The facts If a mixture of methane and chlorine is exposed to a flame, it explodes - producing carbon and hydrogen chloride. This isn't a very useful reaction! The reaction we are going to explore is a more gentle one between methane and chlorine in the presence of ultraviolet light - typically sunlight. This is a good example of a photochemical reaction - a reaction brought about by light. | |||||||||||||
|
Note: These reactions are sometimes described as examples of photocatalysis - reactions catalysed by light. It is better to use the term "photochemical" and keep the keep the word "catalysis" for reactions speeded up by actual substances rather than light. | |||||||||||||
One of the hydrogen atoms in the methane has been replaced by a chlorine atom, so this is a substitution reaction. However, the reaction doesn't stop there, and all the hydrogens in the methane can in turn be replaced by chlorine atoms. Multiple substitution is dealt with on a separate page, and you will find a link to that at the bottom of this page. The mechanism The mechanism involves a chain reaction. During a chain reaction, for every reactive species you start off with, a new one is generated at the end - and this keeps the process going. | |||||||||||||
|
Species: a useful word which is used in chemistry to mean any sort of particle you want it to mean. It covers molecules, ions, atoms, or (in this case) free radicals. | |||||||||||||
|
The over-all process is known as free radical substitution, or as a free radical chain reaction. | |||||||||||||
|
Note: If you aren't sure about the words free radical or substitution, read the page What is free radical substitution? Use the BACK button on your browser to return quickly to this page. | |||||||||||||
|
Chain initiation The chain is initiated (started) by UV light breaking a chlorine molecule into free radicals.
These are the reactions which keep the chain going.
These are reactions which remove free radicals from the system without replacing them by new ones.
| |||||||||||||
Sunday, 30 June 2013
THE REACTION BETWEEN METHANE AND CHLORINE
FREE RADICAL SUBSTITUTION
FREE RADICAL SUBSTITUTION
Substitution reactions
These are reactions in which one atom in a molecule is replaced by
another atom or group of atoms. Free radical substitution for A' level
purposes involves breaking a carbon-hydrogen bond in alkanes such as
It also happens in alkyl groups like methyl, ethyl (and so on) wherever these appear in more complicated molecules.
A simple example of substitution is the reaction between methane and chlorine in the presence of UV light (or sunlight).
Free radical reactions Free radicals are atoms or groups of atoms which have a single unpaired electron. A free radical substitution reaction is one involving these radicals. Free radicals are formed if a bond splits evenly - each atom getting one of the two electrons. The name given to this is homolytic fission. | |||||||||||||
|
Note: If a bond were to split unevenly (one atom getting both electrons, and the other none), ions would be formed. The atom that got both electrons would become negatively charged, while the other one would become positive. This is called heterolytic fission. Warning! It is important that you get these terms the right way round. "Fission" is obvious - it just means "splitting". "Homo" and "hetero" are used in the sense of "same" (homo) or "different" (hetero). This is just like their use in "homosexual" or "heterosexual". So, homolytic fission is splitting a bond to produce two particles which are the same in the sense that they both have a single unpaired electron (both are free radicals). Heterolytic fission produces two particles which are different because one is a positive ion and the other a negative ion. | |||||||||||||
To show that a species (either an atom or a group of atoms) is a free
radical, the symbol is written with a dot attached to show the unpaired
electron. For example:
| |||||||||||||
|
Note: If you wanted to be fussy, the dot showing the electron really ought to be written next to the carbon atom in the methyl radical, because that's the atom with the unpaired electron - in other words as | |||||||||||||
The Basics Nuclear Magnetic Resonance Spectroscopy
Nuclei possessing angular moment (also called spin) have an associated magnetic moment.
A few examples of magnetic isotopes are 13C, 1H, 19F,14N,
17O, 31P, and 33S. Please note that not every isotope is
magnetic. In particular, you should note that 12C is not magnetic. If a nucleus
is not magnetic, it can't be studied by nuclear magnetic resonance spectroscopy. For the
purposes of this course, we will be most interested in 1H and 13C. I
will limit my discussions to 1H in this short treatment. Generally speaking,
you should think of these special nuclei as tiny, atomic, bar magnets.
Nuclear Magnetic Spectroscopy is based on the fact that when a population of magnetic nuclei is placed in an external magnetic field, the nuclei become aligned in a predictable and finite number of orientations. For 1H there are two orientations. In one orientation the protons are aligned with the external magnetic field (north pole of the nucleus aligned with the south pole of the magnet and south pole of the nucleus with the north pole of the magnet) and in the other where the nuclei are aligned against the field (north with north, south with south). The alignment with the field is also called the "alpha" orientation and the alignment against the field is called the "beta" orientation. From my description of the poles, which orientation do you think is the preferred or lower in energy? If you guessed the "alpha", you are correct. It might be worth noting at this point that before the nuclei are placed in the magnetic field they have random orientation
Since the alpha orientation is preferred, more of the population of nuclei are aligned
with the field than against the field. You might wonder why any spins would align against
the field. Realize that we are talking about atomic magnets. These are very, very weak
magnets. The energy difference between the alpha and beta orientations is not large. There
is enough energy for nuclei to exchange between the two orientations at room temperature,
though a slight excess on average is in the lower energy, alpha state.
The nuclear magnetic resonance (NMR) spectroscopy experiment involves using energy in the form of electromagnetic radiation to pump the excess alpha oriented nuclei into the beta state. When the energy is removed, the energized nuclei relax back to the alpha state. The fluctuation of the magnetic field associated with this relaxation process is called resonance and this resonance can be detected and converted into the peaks we see in an NMR spectrum.
What sort of electromagnetic radiation is appropriate for the low energy transition involved in NMR? Well believe it or not, radio waves do the trick. Radio waves are at the very low energy end of the electromagnetic spectrum and are sufficient to induce the desired transition. It is for this reason that NMR is considered to be a safe method of analysis. The same technology is now used in hospitals in MRI (Magnetic Resonance Imagining - people are afraid of the word nuclear). If you have ever had an MRI done, realize that you were placed in a magnetic field and all your magnetic nuclei lined up in the manner described above. Excess nuclei were pumped to higher energy states as you were exposed to radio waves.
The following are two very, very important points to accept and learn if you are going to understand the rest of the discussion.
1. Electric currents have associated magnetic fields.
2. Magnetic fields can generate electric currents.
If you haven't had physics yet, try to accept these two points. Certainly most people have at least heard of electromagnets and if so, you probably have some idea about the first statement.
The following is a very important NMR relationship. This expression relates the external field to the frequency of resonance.
In this equation, n
is frequency, m
is the magnetogyric ratio (not needed for this discussion - a
constant for each nucleus). The big thing to glean from this equation is that the external
field and the frequency are directly proportional. If the external field is larger , the
frequency needed to induce the alpha to beta transition is larger. It follows then that in
a larger field, higher frequency radio waves would be needed to induce the transition.
In this context, it is relevant to note that different nuclear magnetic resonance spectrometers have different magnetic field strengths. For example, the NMR on the first floor of Park Hall has a relatively high field, superconducting magnet. Because the field is high (high enough to erase bank cards and interfere with pacemakers and watches), the frequency range needed to excite protons is relatively high. It is called a 300 MHz (MHz = megahertz, a hertz is a cycle per second - a frequency unit) spectrometer, referring to the excitation frequency. The NMR on the second floor of Park Hall has a much weaker electromagnet associated with it. It is a 60 MHz instrument. Since different NMRs have different operating frequencies, spectra cannot be compared from different machines if they are reported in frequency units. For this reason, the universal ppm (parts per million) units are used in NMR. Please note the following relationship between ppm and frequency. The fact that frequency and ppm are directly proportional is all you need to retain for the future discussion and the course in general.
Now let us use these basic ideas to better understand and interpret NMR spectra.
1. Why do we see peaks? When the excited nuclei in the beta orientation start to relax back down to the alpha orientation, a fluctuating magnetic field is created. This fluctuating field generates a current in a receiver coil that is around the sample. The current is electronically converted into a peak. It is the relaxation that actually gives the peak not the excitation.
2. Why do we see peaks at different positions? Realize that in principle, a peak will be observed for every magnetically distinct nucleus in a molecule. This happens because nuclei that are not in identical structural situations do not experience the external magnetic field to the same extent. The nuclei are shielded or deshielded due to small local fields generated by circulating sigma, pi and lone pair electrons.
To understand this concept better, consider a "run of the mill" hydrogen like that in ethane or methane. When this sort of hydrogen is placed in a magnetic field, the sigma electrons start to circulate. Remember : Magnetic fields generate currents. When the electrons circulate, they generate a small magnetic field that happens to point in the opposite direction to the external field. Remember: Currents have associated magnetic fields. Since magnetism is a vector quantity (vector quantities have direction and magnitude), this local field reduces the overall field somewhat. Therefore, the described hydrogen experiences a reduced magnetic field. If we reconsider the important NMR equation given on page two of this document, we can only conclude that if the external field is lower then the frequency of the electromagnetic radiation needed to induce the alpha to beta transition must be lower. Remember that frequency and ppm are directly proportional. Therefore, if a hydrogen requires a lower frequency, then it will show up as a peak at a lower ppm value. Hydrogens like those in methane are at around 1.0 ppm in the NMR spectrum.
Now consider a hydrogen near a halogen as in bromoethane. This type of hydrogen is in a magnetically altered situation as compared to the hydrogen in methane. Due to its inherent electronegativity, the halogen atom has the effect of pulling sigma electron density away from the hydrogens in the molecule. The effect is largest for the hydrogens closest to the halogen atom. Though the little local opposing sigma field is still generated next to the hydrogens, it is partially pulled away by the electronegative bromine . Therefore, the hydrogens experience less of the local field and more of the external field. In other words, the vector in the vicinity of the hydrogen has been reduced as compared to methane. After you do the vector addition you end up with a larger overall field (again, as compared to methane). So going back to the fact that field and frequency are directly proportional, hydrogens near an electronegative atom should require a higher frequency to flip from the alpha to beta orientation. Therefore, they should appear at a higher ppm in the spectrum. Hydrogens like those in bromoethane should appear from ca. 2.5-4.0 ppm in the NMR spectrum.

Now as a last example, let us consider the NMR spectrum of benzene. Benzene and aromatics in general are very interesting because their hydrogens appear around 7 ppm even though they have no electronegative atoms. Why is this so? It has to do with the pi electrons. Because benzene and its relatives are aromatic, the p orbitals at each carbon in the ring overlap forming one continuous pi system. When the benzene ring is placed in a magnetic field, the external field induces a current in the pi system and that current generates a secondary magnetic field (or induced magnetic field). Once again, remember that electric currents have associated magnetic fields and that magnetic fields generate currents. The secondary magnetic field is such that it adds to the external field in the vicinity of the aromatic hydrogens as diagrammed below.

It is really interesting to consider 18-annulene diagrammed below. 18-Annulene is a large enough ring to have both cis and trans double bonds. This means that some of the hydrogens are pointing in toward the center of the aromatic ring. Reconsider the diagram of benzene above. If you look at it carefully, you will see that the magnetic field opposes the external field on the inside of the ring!!! If 18-annulene is aromatic like benzene, the inner hydrogens should absorb at lower frequency (ppm) and guess what? They do - they appear at -1.7 ppm!! Isn't that neat!!

So summing up, the different hydrogens of a molecule appear at different positions because small local magnetic fields are generated when local electrons begin to circulate due to the effect of the external magnetic field. These small fields either add to or subtract from the external field altering the frequency needed for excitation. Some of the effects are due to the circulation of sigma electrons while others are due to the circulation of pi electrons. The pi effects can be the most dramatic as was demonstrated in the preceding examples.
3. What causes splitting?
Many peaks in NMR spectra appear as symmetric patterns called doublets, triplets, quartets, quintets, etc. When you see these patterns it tells you about the number of adjacent (usually on the carbon next door to that bearing the absorbing hydrogen(s)), but different hydrogens. In simple spectra such as those we will be studying in organic chemistry lab, the number of peaks you see is one more than the number of adjacent, but different hydrogens. This is the so called n+1 rule. Different means that the adjacent hydrogens have a unique magnetic environment and absorb at a distinct frequency compared with the hydrogens in question. For example, consider bromoethane (structure given below).
 Bromoethane has two different types of hydrogens so we expect two absorptions in the
NMR spectrum. One absorption corresponds to the two hydrogens that are closest to the
halogen atom. The other to the hydrogens comprising the methyl group that is farther away.
Based on what I described above with regard to chemical shift (the ppm value), the
hydrogens nearer the bromine should be at a higher ppm position. The hydrogens further
from the bromine should be at lower ppm position.
Bromoethane has two different types of hydrogens so we expect two absorptions in the
NMR spectrum. One absorption corresponds to the two hydrogens that are closest to the
halogen atom. The other to the hydrogens comprising the methyl group that is farther away.
Based on what I described above with regard to chemical shift (the ppm value), the
hydrogens nearer the bromine should be at a higher ppm position. The hydrogens further
from the bromine should be at lower ppm position.
Anyway, getting back the splitting, the hydrogens closer to the bromine will appear as a quartet because they are near three different hydrogens (the hydrogens on the methyl group). Those adjacent hydrogens are communicating their presence to the hydrogens being flipped. They are saying, "We are your neighbors and there are three of us." The reason they are able to communicate their presence is that they are little magnets and as such, they either add to or subtract from the external magnetic field depending on their orientation. Since there are many protons in a sample, the following are the possibilities for the neighboring hydrogens during excitation:
 Please note that in the above diagram the "a" hydrogens are the ones near the
bromine being flipped from the alpha to beta orientation. The "b" hydrogens are
the three neighbors. As shown above, it is possible that a given "a" hydrogen
will have three "b" hydrogens nearby that are aligned with the applied field
during excitation. It is also possible that the three neighbors could all be aligned
against the applied field. More probable is that either two protons will be aligned
against the field or two with the field. These situations are more probable because there
are more combinations of the three nuclei that give rise to these two possibilities. Since
there are three combinations of each of these two, they are each three times more probable
than having all three adjacent nuclei aligned with or against the field.
Please note that in the above diagram the "a" hydrogens are the ones near the
bromine being flipped from the alpha to beta orientation. The "b" hydrogens are
the three neighbors. As shown above, it is possible that a given "a" hydrogen
will have three "b" hydrogens nearby that are aligned with the applied field
during excitation. It is also possible that the three neighbors could all be aligned
against the applied field. More probable is that either two protons will be aligned
against the field or two with the field. These situations are more probable because there
are more combinations of the three nuclei that give rise to these two possibilities. Since
there are three combinations of each of these two, they are each three times more probable
than having all three adjacent nuclei aligned with or against the field.
Now let us think about what these neighboring, local magnets do to the overall field. The "a" hydrogens that have all three neighbors aligned against the field have a lower overall magnetic field. Going back to the fundamental nuclear magnetic resonance equation (see page 2 ), you would conclude that these "a" hydrogens would have a lower frequency requirement for the alpha to beta transition and therefore appear at lower ppm. For the "a" hydrogens having three neighbors with all three "b" hydrogens aligned with the external field, the cumulative local field adds to the external field. This resultant field is larger than the external field so higher frequency electromagnetic radiation is needed to induce the alpha to beta transition. For the "b" hydrogens near two nuclei aligned with the field and one nucleus aligned against the field there is a slight increase in overall field leading to slightly higher frequency requirements. Similarly, two spins aligned against and one aligned with the field leads to slightly lower frequency requirements. So in the end, the "a" population is divided into four groups appearing at slightly different frequencies. The intermediate frequency peaks are taller than the higher and lower frequency peaks because they reflect more probable situations for local hydrogens. Hence, a quartet is observed.
Now if you understand why the "a" hydrogens give a quartet can you figure out why the "b" hydrogens give a triplet? Try to work it out using vectors as done in the above diagram.
For simple systems like bromoethane, n + 1 peaks will be observed for a given absorption, where n = the number of neighboring, but different hydrogens. This formula can be very useful when interpreting simple spectra.
1. The number of peaks. The number of peaks is directly related to symmetry. If a compound has three significantly different types of hydrogens, it should have three different NMR absorptions.
2. The area under each absorption (the integral). The relative areas (or integrals) of the various absorptions in an NMR spectrum equals the relative number of hydrogens absorbing. If we know the molecular formula of a compound, we can use this ratio to figure out the actual number of each type of hydrogen. From the numbers of each type, we can infer the carbon structure. For example, with bromoethane, the relative areas under the NMR peaks are 2:3. This tells us that there is a group of two hydrogens that are the same and another group of three hydrogens that are the same.With your current knowledge of organic chemistry, it seems most likely that the compound has a methyl (-CH3) and a methylene (-CH2-) group. In other words, the most probable way to have three identical hydrogens is on a methyl group. The most probable way to have two identical hydrogens is in a methylene group.
Suppose you have a compound with the formula C5H12O and you are told that there are two NMR peaks, having the relative areas of 1:3. Can you come up with the structure of the compound?
3. The splitting pattern. For this semester, we will be using the n+1 rule as it applies to the simple structures we will be determining. You will see one more peak than the number of adjacent, but different hydrogens. Therefore, you can look at any peak and automatically know how many neighbors there are. This is crucial information because it allows you to start to hook atoms together in your structure. The problem is that people often confuse integral with splitting. So you must always remember this saying "Integral tells you what is here and splitting tells you what is near" This means that the integral tells you about the absorbing hydrogens and the splitting tells you about the neighbors. So what does it mean if you see a quintet with an area of two in a spectrum?
4. The position of the peak or the chemical shift ( ). This tells you about the electronic environment (the electronic environment directly relates to the magnetic environment) of the absorbing hydrogens. It will tell you if there are pi bonds or electronegative atoms nearby, etc. There are nice tables available that organize how different groups effect the frequency of absorptions and in lab you will always have these tables available to you. Yes, you will even have them on exams. A good rule of thumb when you are solving spectra is that the closer a hydrogen is to an electronegative atom the higher the ppm position. This little rule only works if the hydrogen is two or more bonds away from the atom. You will soon see the utility of this when you begin your problems in the workshop. It is also useful to keep in your head that aromatic hydrogens absorb at around 7 ppm.
A few tricks of the trade that are generally useful for spectral problem solving......
1. Always calculate the index of hydrogen deficiency or unsaturation number at the beginning of a problem ( you will normally be given the formula of the compound). Determining the unsaturation number is very helpful in regard to knowing which structural elements need to be present in your final solution. The unsaturation number is where you compare the actual formula with the theoretical saturated formula and compute the number of pairs of hydrogens that are missing. This topic should have been covered in class by now.
2. It is a good idea to interpret your IR spectrum before you do the NMR spectrum so that you have an idea about which functional groups are present in your molecule.
3. Organize your ideas about the structure of the unknown as you go along. For some people it is helpful to set up the following table for the NMR data and conclusions. The important part of the table is the conclusion column in which you are drawing a structural conclusion about the absorbing hydrogens and their neighbors. You should write a structural fragment down as has been done below for bromoethane.
4. You will notice as we do problems in class that we tend to emphasize and draw the
most information from the integral and splitting. Chemical shift (ppm position) in many
cases is the last point of interest. There are a few relevant chemical shifts that should
be interpreted at the outset of a problem.. One is the aromatic chemical shift. Aromatic
hydrogens absorb at ca. 7 ppm. This is a very distinct and characteristic shift and should
be interpreted immediately. If you observe a peak at seven chances are you have an
aromatic ring. The most common aromatic ring is benzene. Another very distinctive shift is
that of the aldehyde functional group. Aldehydic hydrogens appear at ca. 9 ppm in the
spectrum. if you see a shift of nine ppm assume that you have an aldehyde functional
group.
5. Solving spectra rapidly involves making good educated guesses. If you get an integral of three there is really only one probable way to have three identical hydrogens - a methyl group. If you get an integral of nine it is most likely three methyl groups that are the same by symmetry. If you get aromatic absorptions, you probably have one or more benzene rings. Always start with the simplest ideas and work your way toward more exotic solutions.
Nuclear Magnetic Spectroscopy is based on the fact that when a population of magnetic nuclei is placed in an external magnetic field, the nuclei become aligned in a predictable and finite number of orientations. For 1H there are two orientations. In one orientation the protons are aligned with the external magnetic field (north pole of the nucleus aligned with the south pole of the magnet and south pole of the nucleus with the north pole of the magnet) and in the other where the nuclei are aligned against the field (north with north, south with south). The alignment with the field is also called the "alpha" orientation and the alignment against the field is called the "beta" orientation. From my description of the poles, which orientation do you think is the preferred or lower in energy? If you guessed the "alpha", you are correct. It might be worth noting at this point that before the nuclei are placed in the magnetic field they have random orientation
 |
|
random orientation
outside of field |
alpha and beta orientation in field |
The nuclear magnetic resonance (NMR) spectroscopy experiment involves using energy in the form of electromagnetic radiation to pump the excess alpha oriented nuclei into the beta state. When the energy is removed, the energized nuclei relax back to the alpha state. The fluctuation of the magnetic field associated with this relaxation process is called resonance and this resonance can be detected and converted into the peaks we see in an NMR spectrum.
What sort of electromagnetic radiation is appropriate for the low energy transition involved in NMR? Well believe it or not, radio waves do the trick. Radio waves are at the very low energy end of the electromagnetic spectrum and are sufficient to induce the desired transition. It is for this reason that NMR is considered to be a safe method of analysis. The same technology is now used in hospitals in MRI (Magnetic Resonance Imagining - people are afraid of the word nuclear). If you have ever had an MRI done, realize that you were placed in a magnetic field and all your magnetic nuclei lined up in the manner described above. Excess nuclei were pumped to higher energy states as you were exposed to radio waves.
The following are two very, very important points to accept and learn if you are going to understand the rest of the discussion.
1. Electric currents have associated magnetic fields.
2. Magnetic fields can generate electric currents.
If you haven't had physics yet, try to accept these two points. Certainly most people have at least heard of electromagnets and if so, you probably have some idea about the first statement.
The following is a very important NMR relationship. This expression relates the external field to the frequency of resonance.
| n= | mHo |
| 2p |
In this context, it is relevant to note that different nuclear magnetic resonance spectrometers have different magnetic field strengths. For example, the NMR on the first floor of Park Hall has a relatively high field, superconducting magnet. Because the field is high (high enough to erase bank cards and interfere with pacemakers and watches), the frequency range needed to excite protons is relatively high. It is called a 300 MHz (MHz = megahertz, a hertz is a cycle per second - a frequency unit) spectrometer, referring to the excitation frequency. The NMR on the second floor of Park Hall has a much weaker electromagnet associated with it. It is a 60 MHz instrument. Since different NMRs have different operating frequencies, spectra cannot be compared from different machines if they are reported in frequency units. For this reason, the universal ppm (parts per million) units are used in NMR. Please note the following relationship between ppm and frequency. The fact that frequency and ppm are directly proportional is all you need to retain for the future discussion and the course in general.
| Chemical shift in ppm = | peak position in Hz (relative to TMS) |
| spectrometer frequency in MHz |
1. Why do we see peaks? When the excited nuclei in the beta orientation start to relax back down to the alpha orientation, a fluctuating magnetic field is created. This fluctuating field generates a current in a receiver coil that is around the sample. The current is electronically converted into a peak. It is the relaxation that actually gives the peak not the excitation.
2. Why do we see peaks at different positions? Realize that in principle, a peak will be observed for every magnetically distinct nucleus in a molecule. This happens because nuclei that are not in identical structural situations do not experience the external magnetic field to the same extent. The nuclei are shielded or deshielded due to small local fields generated by circulating sigma, pi and lone pair electrons.
To understand this concept better, consider a "run of the mill" hydrogen like that in ethane or methane. When this sort of hydrogen is placed in a magnetic field, the sigma electrons start to circulate. Remember : Magnetic fields generate currents. When the electrons circulate, they generate a small magnetic field that happens to point in the opposite direction to the external field. Remember: Currents have associated magnetic fields. Since magnetism is a vector quantity (vector quantities have direction and magnitude), this local field reduces the overall field somewhat. Therefore, the described hydrogen experiences a reduced magnetic field. If we reconsider the important NMR equation given on page two of this document, we can only conclude that if the external field is lower then the frequency of the electromagnetic radiation needed to induce the alpha to beta transition must be lower. Remember that frequency and ppm are directly proportional. Therefore, if a hydrogen requires a lower frequency, then it will show up as a peak at a lower ppm value. Hydrogens like those in methane are at around 1.0 ppm in the NMR spectrum.
 |
|
| methane | bromoethane |
Now consider a hydrogen near a halogen as in bromoethane. This type of hydrogen is in a magnetically altered situation as compared to the hydrogen in methane. Due to its inherent electronegativity, the halogen atom has the effect of pulling sigma electron density away from the hydrogens in the molecule. The effect is largest for the hydrogens closest to the halogen atom. Though the little local opposing sigma field is still generated next to the hydrogens, it is partially pulled away by the electronegative bromine . Therefore, the hydrogens experience less of the local field and more of the external field. In other words, the vector in the vicinity of the hydrogen has been reduced as compared to methane. After you do the vector addition you end up with a larger overall field (again, as compared to methane). So going back to the fact that field and frequency are directly proportional, hydrogens near an electronegative atom should require a higher frequency to flip from the alpha to beta orientation. Therefore, they should appear at a higher ppm in the spectrum. Hydrogens like those in bromoethane should appear from ca. 2.5-4.0 ppm in the NMR spectrum.
1H NMR Spectrum of Bromoethane
Now as a last example, let us consider the NMR spectrum of benzene. Benzene and aromatics in general are very interesting because their hydrogens appear around 7 ppm even though they have no electronegative atoms. Why is this so? It has to do with the pi electrons. Because benzene and its relatives are aromatic, the p orbitals at each carbon in the ring overlap forming one continuous pi system. When the benzene ring is placed in a magnetic field, the external field induces a current in the pi system and that current generates a secondary magnetic field (or induced magnetic field). Once again, remember that electric currents have associated magnetic fields and that magnetic fields generate currents. The secondary magnetic field is such that it adds to the external field in the vicinity of the aromatic hydrogens as diagrammed below.
Benzene
If the local field is in the same direction as the external field, the resulting field
is larger than the external field. This means that the frequency needed to flip those
hydrogens experiencing that field is larger. Larger frequency translates into higher ppm
position.It is really interesting to consider 18-annulene diagrammed below. 18-Annulene is a large enough ring to have both cis and trans double bonds. This means that some of the hydrogens are pointing in toward the center of the aromatic ring. Reconsider the diagram of benzene above. If you look at it carefully, you will see that the magnetic field opposes the external field on the inside of the ring!!! If 18-annulene is aromatic like benzene, the inner hydrogens should absorb at lower frequency (ppm) and guess what? They do - they appear at -1.7 ppm!! Isn't that neat!!
[18]-Annulene
So summing up, the different hydrogens of a molecule appear at different positions because small local magnetic fields are generated when local electrons begin to circulate due to the effect of the external magnetic field. These small fields either add to or subtract from the external field altering the frequency needed for excitation. Some of the effects are due to the circulation of sigma electrons while others are due to the circulation of pi electrons. The pi effects can be the most dramatic as was demonstrated in the preceding examples.
3. What causes splitting?
Many peaks in NMR spectra appear as symmetric patterns called doublets, triplets, quartets, quintets, etc. When you see these patterns it tells you about the number of adjacent (usually on the carbon next door to that bearing the absorbing hydrogen(s)), but different hydrogens. In simple spectra such as those we will be studying in organic chemistry lab, the number of peaks you see is one more than the number of adjacent, but different hydrogens. This is the so called n+1 rule. Different means that the adjacent hydrogens have a unique magnetic environment and absorb at a distinct frequency compared with the hydrogens in question. For example, consider bromoethane (structure given below).
Anyway, getting back the splitting, the hydrogens closer to the bromine will appear as a quartet because they are near three different hydrogens (the hydrogens on the methyl group). Those adjacent hydrogens are communicating their presence to the hydrogens being flipped. They are saying, "We are your neighbors and there are three of us." The reason they are able to communicate their presence is that they are little magnets and as such, they either add to or subtract from the external magnetic field depending on their orientation. Since there are many protons in a sample, the following are the possibilities for the neighboring hydrogens during excitation:
Now let us think about what these neighboring, local magnets do to the overall field. The "a" hydrogens that have all three neighbors aligned against the field have a lower overall magnetic field. Going back to the fundamental nuclear magnetic resonance equation (see page 2 ), you would conclude that these "a" hydrogens would have a lower frequency requirement for the alpha to beta transition and therefore appear at lower ppm. For the "a" hydrogens having three neighbors with all three "b" hydrogens aligned with the external field, the cumulative local field adds to the external field. This resultant field is larger than the external field so higher frequency electromagnetic radiation is needed to induce the alpha to beta transition. For the "b" hydrogens near two nuclei aligned with the field and one nucleus aligned against the field there is a slight increase in overall field leading to slightly higher frequency requirements. Similarly, two spins aligned against and one aligned with the field leads to slightly lower frequency requirements. So in the end, the "a" population is divided into four groups appearing at slightly different frequencies. The intermediate frequency peaks are taller than the higher and lower frequency peaks because they reflect more probable situations for local hydrogens. Hence, a quartet is observed.
Now if you understand why the "a" hydrogens give a quartet can you figure out why the "b" hydrogens give a triplet? Try to work it out using vectors as done in the above diagram.
For simple systems like bromoethane, n + 1 peaks will be observed for a given absorption, where n = the number of neighboring, but different hydrogens. This formula can be very useful when interpreting simple spectra.
The Interpretation of Simple NMR Spectra
This year, we will abstract the following information from NMR spectra to determine
structures of products from organic reactions and isolations. 1. The number of peaks. The number of peaks is directly related to symmetry. If a compound has three significantly different types of hydrogens, it should have three different NMR absorptions.
2. The area under each absorption (the integral). The relative areas (or integrals) of the various absorptions in an NMR spectrum equals the relative number of hydrogens absorbing. If we know the molecular formula of a compound, we can use this ratio to figure out the actual number of each type of hydrogen. From the numbers of each type, we can infer the carbon structure. For example, with bromoethane, the relative areas under the NMR peaks are 2:3. This tells us that there is a group of two hydrogens that are the same and another group of three hydrogens that are the same.With your current knowledge of organic chemistry, it seems most likely that the compound has a methyl (-CH3) and a methylene (-CH2-) group. In other words, the most probable way to have three identical hydrogens is on a methyl group. The most probable way to have two identical hydrogens is in a methylene group.
Suppose you have a compound with the formula C5H12O and you are told that there are two NMR peaks, having the relative areas of 1:3. Can you come up with the structure of the compound?
3. The splitting pattern. For this semester, we will be using the n+1 rule as it applies to the simple structures we will be determining. You will see one more peak than the number of adjacent, but different hydrogens. Therefore, you can look at any peak and automatically know how many neighbors there are. This is crucial information because it allows you to start to hook atoms together in your structure. The problem is that people often confuse integral with splitting. So you must always remember this saying "Integral tells you what is here and splitting tells you what is near" This means that the integral tells you about the absorbing hydrogens and the splitting tells you about the neighbors. So what does it mean if you see a quintet with an area of two in a spectrum?
4. The position of the peak or the chemical shift ( ). This tells you about the electronic environment (the electronic environment directly relates to the magnetic environment) of the absorbing hydrogens. It will tell you if there are pi bonds or electronegative atoms nearby, etc. There are nice tables available that organize how different groups effect the frequency of absorptions and in lab you will always have these tables available to you. Yes, you will even have them on exams. A good rule of thumb when you are solving spectra is that the closer a hydrogen is to an electronegative atom the higher the ppm position. This little rule only works if the hydrogen is two or more bonds away from the atom. You will soon see the utility of this when you begin your problems in the workshop. It is also useful to keep in your head that aromatic hydrogens absorb at around 7 ppm.
A few tricks of the trade that are generally useful for spectral problem solving......
1. Always calculate the index of hydrogen deficiency or unsaturation number at the beginning of a problem ( you will normally be given the formula of the compound). Determining the unsaturation number is very helpful in regard to knowing which structural elements need to be present in your final solution. The unsaturation number is where you compare the actual formula with the theoretical saturated formula and compute the number of pairs of hydrogens that are missing. This topic should have been covered in class by now.
2. It is a good idea to interpret your IR spectrum before you do the NMR spectrum so that you have an idea about which functional groups are present in your molecule.
3. Organize your ideas about the structure of the unknown as you go along. For some people it is helpful to set up the following table for the NMR data and conclusions. The important part of the table is the conclusion column in which you are drawing a structural conclusion about the absorbing hydrogens and their neighbors. You should write a structural fragment down as has been done below for bromoethane.
| ppm | integral | splitting | conclusion |
| 1.6 | 3 | triplet | CH3CH2- |
| 3.4 | 2 | quartet | -CH2CH3 near electronegative atom |
5. Solving spectra rapidly involves making good educated guesses. If you get an integral of three there is really only one probable way to have three identical hydrogens - a methyl group. If you get an integral of nine it is most likely three methyl groups that are the same by symmetry. If you get aromatic absorptions, you probably have one or more benzene rings. Always start with the simplest ideas and work your way toward more exotic solutions.
Spectroscopy Types
Absorption
Absorption spectroscopy is a technique in which the power of a beam of light measured before and after interaction with a sample is compared. Specific absorption techniques tend to be referred to by the wavelength of radiation measured such as ultraviolet, infrared or microwave absorption spectroscopy. Absorption occurs when the energy of the photons matches the energy difference between two states of the material.Fluorescence
Fluorescence spectroscopy uses higher energy photons to excite a sample, which will then emit lower energy photons. This technique has become popular for its biochemical and medical applications, and can be used for confocal microscopy, fluorescence resonance energy transfer, and fluorescence lifetime imaging.X-ray
When X-rays of sufficient frequency (energy) interact with a substance, inner shell electrons in the atom are excited to outer empty orbitals, or they may be removed completely, ionizing the atom. The inner shell "hole" will then be filled by electrons from outer orbitals. The energy available in this de-excitation process is emitted as radiation (fluorescence) or will remove other less-bound electrons from the atom (Auger effect). The absorption or emission frequencies (energies) are characteristic of the specific atom. In addition, for a specific atom, small frequency (energy) variations that are characteristic of the chemical bonding occur. With a suitable apparatus, these characteristic X-ray frequencies or Auger electron energies can be measured. X-ray absorption and emission spectroscopy is used in chemistry and material sciences to determine elemental composition and chemical bonding.X-ray crystallography is a scattering process; crystalline materials scatter X-rays at well-defined angles. If the wavelength of the incident X-rays is known, this allows calculation of the distances between planes of atoms within the crystal. The intensities of the scattered X-rays give information about the atomic positions and allow the arrangement of the atoms within the crystal structure to be calculated. However, the X-ray light is then not dispersed according to its wavelength, which is set at a given value, and X-ray diffraction is thus not a spectroscopy.
Flame
Liquid solution samples are aspirated into a burner or nebulizer/burner combination, desolvated, atomized, and sometimes excited to a higher energy electronic state. The use of a flame during analysis requires fuel and oxidant, typically in the form of gases. Common fuel gases used are acetylene (ethyne) or hydrogen. Common oxidant gases used are oxygen, air, or nitrous oxide. These methods are often capable of analyzing metallic element analytes in the part per million, billion, or possibly lower concentration ranges. Light detectors are needed to detect light with the analysis information coming from the flame.- Atomic Emission Spectroscopy - This method uses flame excitation; atoms are excited from the heat of the flame to emit light. This method commonly uses a total consumption burner with a round burning outlet. A higher temperature flame than atomic absorption spectroscopy (AA) is typically used to produce excitation of analyte atoms. Since analyte atoms are excited by the heat of the flame, no special elemental lamps to shine into the flame are needed. A high resolution polychromator can be used to produce an emission intensity vs. wavelength spectrum over a range of wavelengths showing multiple element excitation lines, meaning multiple elements can be detected in one run. Alternatively, a monochromator can be set at one wavelength to concentrate on analysis of a single element at a certain emission line. Plasma emission spectroscopy is a more modern version of this method. See Flame emission spectroscopy for more details.
- Atomic absorption spectroscopy (often called AA) - This method commonly uses a pre-burner nebulizer (or nebulizing chamber) to create a sample mist and a slot-shaped burner that gives a longer pathlength flame. The temperature of the flame is low enough that the flame itself does not excite sample atoms from their ground state. The nebulizer and flame are used to desolvate and atomize the sample, but the excitation of the analyte atoms is done by the use of lamps shining through the flame at various wavelengths for each type of analyte. In AA, the amount of light absorbed after going through the flame determines the amount of analyte in the sample. A graphite furnace for heating the sample to desolvate and atomize is commonly used for greater sensitivity. The graphite furnace method can also analyze some solid or slurry samples. Because of its good sensitivity and selectivity, it is still a commonly used method of analysis for certain trace elements in aqueous (and other liquid) samples.
- Atomic Fluorescence Spectroscopy - This method commonly uses a burner with a round burning outlet. The flame is used to solvate and atomize the sample, but a lamp shines light at a specific wavelength into the flame to excite the analyte atoms in the flame. The atoms of certain elements can then fluoresce emitting light in a different direction. The intensity of this fluorescing light is used for quantifying the amount of analyte element in the sample. A graphite furnace can also be used for atomic fluorescence spectroscopy. This method is not as commonly used as atomic absorption or plasma emission spectroscopy.
In some ways similar to flame atomic emission spectroscopy, it has largely replaced it.
- Direct-current plasma (DCP)
A plasma support gas is necessary, and Ar is common. Samples can be deposited on one of the electrodes, or if conducting can make up one electrode.
- Glow discharge-optical emission spectrometry (GD-OES)
- Inductively coupled plasma-atomic emission spectrometry (ICP-AES)
- Laser Induced Breakdown Spectroscopy (LIBS), also called Laser-induced plasma spectrometry (LIPS)
- Microwave-induced plasma (MIP)
Nowadays, the spark sources with controlled discharges under an argon atmosphere allow that this method can be considered eminently quantitative, and its use is widely expanded worldwide through production control laboratories of foundries and steel mills.
Visible
Many atoms emit or absorb visible light. In order to obtain a fine line spectrum, the atoms must be in a gas phase. This means that the substance has to be vaporised. The spectrum is studied in absorption or emission. Visible absorption spectroscopy is often combined with UV absorption spectroscopy in UV/Vis spectroscopy. Although this form may be uncommon as the human eye is a similar indicator, it still proves useful when distinguishing colours.Ultraviolet
All atoms absorb in the Ultraviolet (UV) region because these photons are energetic enough to excite outer electrons. If the frequency is high enough, photoionization takes place. UV spectroscopy is also used in quantifying protein and DNA concentration as well as the ratio of protein to DNA concentration in a solution. Several amino acids usually found in protein, such as tryptophan, absorb light in the 280 nm range and DNA absorbs light in the 260 nm range. For this reason, the ratio of 260/280 nm absorbance is a good general indicator of the relative purity of a solution in terms of these two macromolecules. Reasonable estimates of protein or DNA concentration can also be made this way using Beer's law.Infrared
Infrared spectroscopy offers the possibility to measure different types of inter atomic bond vibrations at different frequencies. Especially in organic chemistry the analysis of IR absorption spectra shows what type of bonds are present in the sample. It is also an important method for analysing polymers and constituents like fillers, pigments and plasticizers.Near Infrared (NIR)
The near infrared NIR range, immediately beyond the visible wavelength range, is especially important for practical applications because of the much greater penetration depth of NIR radiation into the sample than in the case of mid IR spectroscopy range. This allows also large samples to be measured in each scan by NIR spectroscopy, and is currently employed for many practical applications such as: rapid grain analysis, medical diagnosis pharmaceuticals/medicines, biotechnology, genomics analysis, proteomic analysis, interactomics research, inline textile monitoring, food analysis and chemical imaging/hyperspectral imaging of intact organisms, plastics, textiles, insect detection, forensic lab application, crime detection and various military applications.Raman
Raman spectroscopy uses the inelastic scattering of light to analyse vibrational and rotational modes of molecules. The resulting 'fingerprints' are an aid to analysis.Coherent anti-Stokes Raman spectroscopy (CARS)
CARS is a recent technique that has high sensitivity and powerful applications for ''in vivo'' spectroscopy and imaging.Nuclear magnetic resonance
Nuclear magnetic resonance spectroscopy analyzes the magnetic properties of certain atomic nuclei to determine different electronic local environments of hydrogen, carbon, or other atoms in an organic compound or other compound. This is used to help determine the structure of the compound.Photoemission
Mössbauer
Transmission or conversion-electron (CEMS) modes of Mössbauer spectroscopy probe the properties of specific isotope nuclei in different atomic environments by analyzing the resonant absorption of characteristic energy gamma-rays known as the Mössbauer effect.Physical Properties of Organic Compounds
Physical Properties of Organic Compounds
Boiling Point and Vapor Pressure
The transition between liquid and gas is very important for organic liquids. It is useful for purification through distillation, and is responsible for evaporation. Finally, without some vapor pressure, no compound could reach your nose to produce those wonderful or terrible smells we associate with organic chemistry.The vapor pressure is the actual pressure of vapor over a liquid. Measurable at all temperatures, it is smaller at lower temperatures, and increases with higher temperatures. Eventually, the vapor pressure will meet or exceed atmospheric pressure, and bubbles will form in the liquid—a process we call “boiling.” The temperature at which the vapor pressure equals atmospheric pressure is the official boiling point.
Let’s think about the physical process involved in boiling. The figure below shows molecules (represented by “X”) in the liquid on the left, and in the gas on the right:
Physical process:
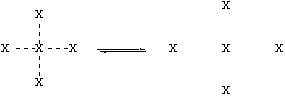
X--X is composite of polar, H-bonding, and Van der Waals forces.
The big change on boiling is that the intermolecular forces (X---X) are broken, and not replaced by anything. [Note, however, that the molecule X stays intact, and the covalent bonds are not broken.] This means that the stronger the intermolecular forces (X—X) are, the harder it will be to disrupt them. This will yield a lower vapor pressure at a given temperature, and higher boiling point. These intermolecular forces will be a composite of polar, H-bonding, and Van der Waals forces.
Predicting Boiling Points
Even for professional chemists, it is very difficult to predict the boiling point of a single compound. However, it is not too difficult in simple cases to predict the relative boiling points of two compounds (that is, which compound boils at the higher temperature).| Boiling points of homologous series of compounds with 8 functional groups. Note the trend towards higher boiling points as compounds get longer within a series, and the four classes of functional groups by strength of intermolecular interaction. | 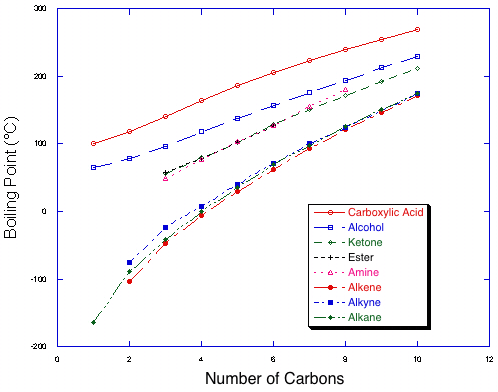 |
Within a series, there is a steady increase in boiling points as compounds get larger. This is due to an increase in surface area, and therefore in van der Waals interactions. The addition of one or two carbons will not cause much of a rise in boiling point, but it will be noticeable and can add up with large changes in size. Note also that the lines get somewhat closer as the molecules get larger, because as one adds more alkane content to the molecule, the molecules increasingly resemble alkanes.
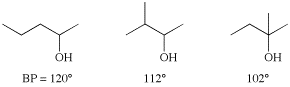
Another important factor in boiling point prediction relates to the difference between branched and linear carbon skeletons. Since branching allows the compound to be more compact, it reduces surface area, reducing the van der Waals interactions, and therefore reducing the boiling point. Note the effects of branching on the following three alcohols:
Saturday, 29 June 2013
THE EXTRACTION OF METALS - AN INTRODUCTION
|
THE EXTRACTION OF METALS - AN INTRODUCTION
This page looks at the various factors which influence the choice of
method for extracting metals from their ores, including reduction by
carbon, reduction by a reactive metal (like sodium or magnesium), and by
electrolysis. Details for the extraction of aluminium, copper, iron and titanium are given in separate pages in this section. From ore to metal What are "ores"? An ore is any naturally-occurring source of a metal that you can economically extract the metal from. Aluminium, for example, is the most common metal in the Earth's crust, occurring in all sorts of minerals. However, it isn't economically worthwhile to extract it from most of these minerals. Instead, the usual ore of aluminium is bauxite - which contains from 50 - 70% of aluminium oxide. | |||||||||||
|
Note: We always treat bauxite as if it was aluminium oxide for chemistry purposes, although it is actually more complicated than that in reality. | |||||||||||
|
Copper is much rarer, but fortunately can be found in high-grade ores
(ones containing a high percentage of copper) in particular places.
Because copper is a valuable metal, it is also worth extracting it from
low-grade ores as well. Ores are commonly oxides - for example:
Concentrating the ore This simply means getting rid of as much of the unwanted rocky material as possible before the ore is converted into the metal. In some cases this is done chemically. For example, pure aluminium oxide is obtained from bauxite by a process involving a reaction with sodium hydroxide solution. This is described in detail on the aluminium page in this section. Some copper ores can be converted into copper(II) sulphate solution by leaving the crushed ore in contact with dilute sulphuric acid for a long time. Copper can then be extracted from the copper(II) sulphate solution. But, in many cases, it is possible to separate the metal compound from unwanted rocky material by physical means. A common example of this involves froth flotation. Froth flotation The ore is first crushed and then treated with something which will bind to the particles of the metal compound that you want and make those particles hydrophobic. "Hydrophobic" literally means "water fearing". In concentrating copper ores, for example, pine oil is often used. The pine oil binds to the copper compounds, but not to the unwanted rocky material. The treated ore is then put in a large bath of water containing a foaming agent (a soap or detergent of some kind), and air is blown through the mixture to make a lot of bubbles. Because they are water-repellent, the coated particles of the metal compound tend to be picked up by the air bubbles, float to the top of the bath, and are allowed to flow out over the sides. The rest of the rocky material stays in the bath. Reducing the metal compound to the metal Why is this reduction? At its simplest, where you are starting from metal oxides, the ore is being reduced because oxygen is being removed. 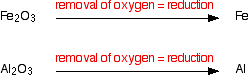 However, if you are starting with a sulphide ore, for example, that's not a lot of help! It is much more helpful to use the definition of reduction in terms of addition of electrons. To a reasonable approximation, you can think of these ores as containing positive metal ions. To convert them to the metal, you need to add electrons - reduction.  | |||||||||||
|
Note: In some compounds the metal may not literally be present as a positive ion. Instead, it may be part of a covalent bond - but will always be the least electronegative element present, and so will carry some degree of positive charge. That means that its oxidation state will always be positive. Reducing that oxidation state to zero (in the raw element) will always involve adding electrons. If you aren't sure about oxidation states you could follow this link to find out about them. If you choose to follow this link, use the BACK button on your browser to return to this page later. | |||||||||||
|
Choosing a method of reduction There are various economic factors you need to think about in choosing a method of reduction for a particular ore. These are all covered in detail on other pages in this section under the extractions of particular metals. What follows is a quick summary. You need to consider:
Carbon reduction Carbon (as coke or charcoal) is cheap. It not only acts as a reducing agent, but it also acts as the fuel to provide heat for the process. However, in some cases (for example with aluminium) the temperature needed for carbon reduction is too high to be economic - so a different method has to be used. Carbon may also be left in the metal as an impurity. Sometimes this can be removed afterwards (for example, in the extraction of iron); sometimes it can't (for example in producing titanium), and a different method would have to be used in cases like this. Reduction using a more reactive metal Titanium is produced by reducing titanium(IV) chloride using a more reactive metal such as sodium or magnesium. As you will see if you read the page about titanium extraction, this is the only way of producing high purity metal. The more reactive metal sodium releases electrons easily as it forms its ions: These electrons are used to reduce the titanium(IV) chloride: | |||||||||||
|
Note: This is a good example of a reduction in which the metal isn't originally present as an ion. Titanium(IV) chloride is a covalent liquid. The reduction is from titanium in the +4 oxidation state to the metal in the zero oxidation state. | |||||||||||
|
The downside of this is expense. You have first to extract (or to
buy) the sodium or magnesium. The more reactive the metal is, the more
difficult and expensive the extraction becomes. That means that you are
having to use a very expensive reducing agent to extract the titanium. As you will see if you read the page about titanium extraction, there are other problems in its extraction which also add to the cost. Reduction by electrolysis This is a common extraction process for the more reactive metals - for example, for aluminium and metals above it in the electrochemical series. You may also come across it in other cases such as one method of extracting copper and in the purification of copper. During electrolysis, electrons are being added directly to the metal ions at the cathode (the negative electrode). The downside (particularly in the aluminium case) is the cost of the electricity. An advantage is that it can produce very pure metals. | |||||||||||
THE GENERAL FEATURES OF TRANSITION METAL CHEMISTRY
|
THE GENERAL FEATURES OF TRANSITION METAL CHEMISTRY
This page explains what a transition metal is in terms of its
electronic structure, and then goes on to look at the general features
of transition metal chemistry. These include variable oxidation state
(oxidation number), complex ion formation, coloured ions, and catalytic
activity. You will find some of this covered quite briefly on this page with links to other parts of the site where the topics are covered in more detail. The electronic structures of transition metals What is a transition metal? The terms transition metal (or element) and d block element are sometimes used as if they mean the same thing. They don't - there's a subtle difference between the two terms. We'll explore d block elements first: d block elements You will remember that when you are building the Periodic Table and working out where to put the electrons using the Aufbau Principle, something odd happens after argon. At argon, the 3s and 3p levels are full, but rather than fill up the 3d levels next, the 4s level fills instead to give potassium and then calcium. Only after that do the 3d levels fill. | |||||||||||||||||||||||||||||||
|
Note: If you aren't sure about atomic orbitals and electronic structures, you really need to follow this link before you go on. It takes you to a page explaining atomic orbitals and then on to other pages about electronic structures. If you do follow the link, use the BACK button on your browser (or the History file or Go menu) to return quickly to this page. | |||||||||||||||||||||||||||||||
The elements in the Periodic Table which correspond to the d levels filling are called d block elements. The first row of these is shown in the shortened form of the Periodic Table below.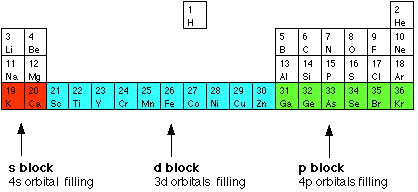
| |||||||||||||||||||||||||||||||
|
Note: This is something that you are just going to have to accept. There is no simple explanation for it which is usable at this level. Any simple explanation which is given is faulty! People sometimes say that a half-filled d level as in chromium (with one electron in each orbital) is stable, and so it is - sometimes! But you then have to look at why it is stable. The obvious explanation is that chromium takes up this structure because separating the electrons minimises the repulsions between them - otherwise it would take up some quite different structure. But you only have to look at the electronic configuration of tungsten (W) to see that this apparently simple explanation doesn't always work. Tungsten has the same number of outer electrons as chromium, but its outer structure is different - 5d46s2. Again the electron repulsions must be minimised - otherwise it wouldn't take up this configuration. But in this case, it isn't true that the half-filled state is the most stable - it doesn't seem very reasonable, but it's a fact! The real explanation is going to be much more difficult than it seems at first sight. Neither can you use the statement that a full d level (for example, in the copper case) is stable, unless you can come up with a proper explanation of why that is. You can't assume that looking nice and tidy is a good enough reason! If you can't explain something properly, it is much better just to accept it than to make up faulty explanations which sound OK on the surface but don't stand up to scrutiny! | |||||||||||||||||||||||||||||||
|
Transition metals Not all d block elements count as transition metals! There are discrepancies between the various UK-based syllabuses, but the majority use the definition:
| |||||||||||||||||||||||||||||||
|
Note: The most recent IUPAC definition includes the possibility of the element itself having incomplete d orbitals as well. This is unlikely to be a big problem (it only really arises with scandium), but it would pay you to learn the version your syllabus wants. Both versions of the definition are currently in use in various UK-based syllabuses. If you are working towards a UK-based exam and haven't got a copy of your syllabus, follow this link to find out how to get one. Use the BACK button on your browser to return quickly to this page. | |||||||||||||||||||||||||||||||
|
On the basis of the definition outlined above, scandium and zinc
don't count as transition metals - even though they are members of the d
block. Scandium has the electronic structure [Ar] 3d14s2. When it forms ions, it always loses the 3 outer electrons and ends up with an argon structure. The Sc3+ ion has no d electrons and so doesn't meet the definition. Zinc has the electronic structure [Ar] 3d104s2. When it forms ions, it always loses the two 4s electrons to give a 2+ ion with the electronic structure [Ar] 3d10. The zinc ion has full d levels and doesn't meet the definition either. By contrast, copper, [Ar] 3d104s1, forms two ions. In the Cu+ ion the electronic structure is [Ar] 3d10. However, the more common Cu2+ ion has the structure [Ar] 3d9. Copper is definitely a transition metal because the Cu2+ ion has an incomplete d level. Transition metal ions Here you are faced with one of the most irritating facts in chemistry at this level! When you work out the electronic structures of the first transition series (from scandium to zinc) using the Aufbau Principle, you do it on the basis that the 3d orbitals have a higher energy than the 4s orbital. That means that you work on the assumption that the 3d electrons are added after the 4s ones. However, in all the chemistry of the transition elements, the 4s orbital behaves as the outermost, highest energy orbital. When these metals form ions, the 4s electrons are always lost first. You must remember this:
| |||||||||||||||||||||||||||||||
|
Note: The problem here is that the Aufbau Principle can only really be used as a way of working out the electronic structures of most atoms. It is a simple way of doing that, although it fails with some, like chromium or copper, of course, and you have to learn these. There is, however, a flaw in the theory behind it which produces problems like this. Why are the apparently higher energy 3d electrons not the ones to get lost when the metal ionises? I have written a detailed explanation of this on another page called the order of filling 3d and 4s orbitals. If you are a teacher or a very confident student then you might like to follow this link. If you aren't so confident, I suggest that you ignore it. Make sure that you can work out the structures of these atoms using the Aufbau Principle on the assumption that the 3d orbitals fill after the 4s, and learn that when the atoms ionise, the 4s electrons are always lost first. Just ignore the contradictions between these two ideas! | |||||||||||||||||||||||||||||||
To write the electronic structure for Co2+:
To write the electronic structure for V3+:
| |||||||||||||||||||||||||||||||
|
Note: You will find more examples of writing the electronic structures for d block ions, by following this link. Use the BACK button on your browser to return quickly to this page. | |||||||||||||||||||||||||||||||
|
Variable oxidation state (number)
One of the key features of transition metal chemistry is the wide
range of oxidation states (oxidation numbers) that the metals can show. | |||||||||||||||||||||||||||||||
|
Note: If you aren't sure about oxidation states, you really need to follow this link before you go on. Use the BACK button on your browser to return quickly to this page. | |||||||||||||||||||||||||||||||
|
It would be wrong, though, to give the impression that only
transition metals can have variable oxidation states. For example,
elements like sulphur or nitrogen or chlorine have a very wide range of
oxidation states in their compounds - and these obviously aren't
transition metals. However, this variability is less common in metals apart from the transition elements. Of the familiar metals from the main groups of the Periodic Table, only lead and tin show variable oxidation state to any extent. Examples of variable oxidation states in the transition metals Iron Iron has two common oxidation states (+2 and +3) in, for example, Fe2+ and Fe3+. It also has a less common +6 oxidation state in the ferrate(VI) ion, FeO42-. Manganese Manganese has a very wide range of oxidation states in its compounds. For example:
You will find the above examples and others looked at in detail if you explore the chemistry of individual metals from the transition metal menu. There is a link to this menu at the bottom of the page. Explaining the variable oxidation states in the transition metals We'll look at the formation of simple ions like Fe2+ and Fe3+. When a metal forms an ionic compound, the formula of the compound produced depends on the energetics of the process. On the whole, the compound formed is the one in which most energy is released. The more energy released, the more stable the compound. There are several energy terms to think about, but the key ones are:
But off-setting this, the more highly charged the ion, the more energy is released either as lattice enthalpy or the hydration enthalpy of the metal ion. | |||||||||||||||||||||||||||||||
|
Note: What I am talking about here in a general way are Born-Haber cycles. You will find these covered in the energetics section of Chemguide, or my chemistry calculations book. | |||||||||||||||||||||||||||||||
|
Thinking about a typical non-transition metal (calcium) Calcium chloride is CaCl2. Why is that? If you tried to make CaCl, (containing a Ca+ ion), the overall process is slightly exothermic. By making a Ca2+ ion instead, you have to supply more ionisation energy, but you get out lots more lattice energy. There is much more attraction between chloride ions and Ca2+ ions than there is if you only have a 1+ ion. The overall process is very exothermic. Because the formation of CaCl2 releases much more energy than making CaCl, then CaCl2 is more stable - and so forms instead. What about CaCl3? This time you have to remove yet another electron from calcium. The first two come from the 4s level. The third one comes from the 3p. That is much closer to the nucleus and therefore much more difficult to remove. There is a large jump in ionisation energy between the second and third electron removed. Although there will be a gain in lattice enthalpy, it isn't anything like enough to compensate for the extra ionisation energy, and the overall process is very endothermic. It definitely isn't energetically sensible to make CaCl3! Thinking about a typical transition metal (iron) Here are the changes in the electronic structure of iron to make the 2+ or the 3+ ion.
The figures for the first three ionisation energies (in kJ mol-1) for iron compared with those of calcium are:
In the iron case, the extra ionisation energy is compensated more or less by the extra lattice enthalpy or hydration enthalpy evolved when the 3+ compound is made. The net effect of all this is that the overall enthalpy change isn't vastly different whether you make, say, FeCl2 or FeCl3. That means that it isn't too difficult to convert between the two compounds. The formation of complex ions What is a complex ion? A complex ion has a metal ion at its centre with a number of other molecules or ions surrounding it. These can be considered to be attached to the central ion by co-ordinate (dative covalent) bonds. (In some cases, the bonding is actually more complicated than that.) The molecules or ions surrounding the central metal ion are called ligands. Simple ligands include water, ammonia and chloride ions.  Some examples of complex ions formed by transition metals
| |||||||||||||||||||||||||||||||
|
Note: You will find much more about complex ions by following this link. It will take you to a part of the site dealing exclusively with complex ions. If you do follow the link, use the BACK button on your browser (or the History file or Go menu) if you want to return to this page again. | |||||||||||||||||||||||||||||||
|
The formation of coloured compounds
Some common examples The diagrams show aproximate colours for some common transition metal complex ions. 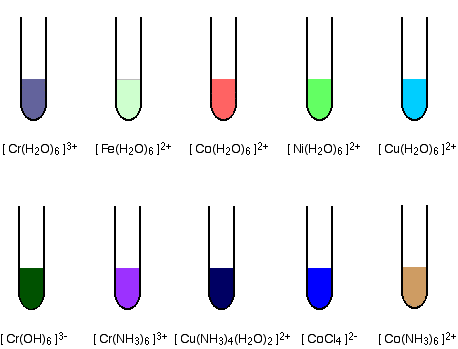 Alternatively, you could explore the complex ions menu (follow the link in the help box which has just disappeared off the top of the screen). The origin of colour in the transition metal ions When white light passes through a solution of one of these ions, or is reflected off it, some colours in the light are absorbed. The colour you see is how your eye perceives what is left. Attaching ligands to a metal ion has an effect on the energies of the d orbitals. Light is absorbed as electrons move between one d orbital and another. This is explained in detail on another page. | |||||||||||||||||||||||||||||||
|
Note: You will find a detailed explanation of the origin of colour in complex ions and the factors which cause it to change by following this link. That page is on the part of the site dealing with complex ions. Use the BACK button on your browser if you want to return to this page again. | |||||||||||||||||||||||||||||||
|
Catalytic activity
Transition metals and their compounds are often good catalysts. A
few of the more obvious cases are mentioned below, but you will find
catalysis explored in detail elsewhere on the site (follow the link
after the examples). Transition metals and their compounds function as catalysts either because of their ability to change oxidation state or, in the case of the metals, to adsorb other substances on to their surface and activate them in the process. All this is expored in the main catalysis section. Transition metals as catalysts Iron in the Haber Process The Haber Process combines hydrogen and nitrogen to make ammonia using an iron catalyst. Nickel in the hydrogenation of C=C bonds This reaction is at the heart of the manufacture of margarine from vegetable oils. However, the simplest example is the reaction between ethene and hydrogen in the presence of a nickel catalyst. Transition metal compounds as catalysts Vanadium(V) oxide in the Contact Process At the heart of the Contact Process is a reaction which converts sulphur dioxide into sulphur trioxide. Sulphur dioxide gas is passed together with air (as a source of oxygen) over a solid vanadium(V) oxide catalyst. Iron ions in the reaction between persulphate ions and iodide ions Persulphate ions (peroxodisulphate ions), S2O82-, are very powerful oxidising agents. Iodide ions are very easily oxidised to iodine. And yet the reaction between them in solution in water is very slow. The reaction is catalysed by the presence of either iron(II) or iron(III) ions. | |||||||||||||||||||||||||||||||
|
| |||||||||||||||||||||||||||||||
Subscribe to:
Posts (Atom)




