Heterocyclic Compounds
Compounds classified as heterocyclic probably constitute the largest and most varied family of organic compounds. After all, every carbocyclic compound, regardless of structure and functionality, may in principle be converted into a collection of heterocyclic analogs by replacing one or more of the ring carbon atoms with a different element. Even if we restrict our consideration to oxygen, nitrogen and sulfur (the most common heterocyclic elements), the permutations and combinations of such a replacement are numerous.Nomenclature
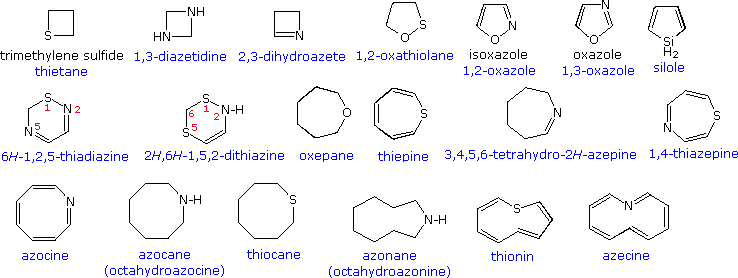
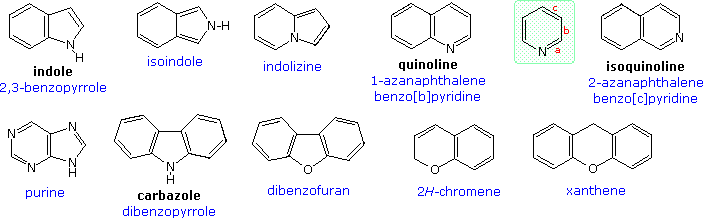
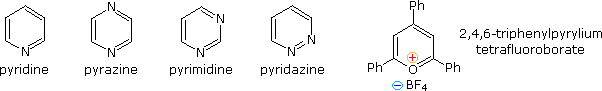
Devising a systematic nomenclature system for heterocyclic compounds presented a formidable challenge, which has not been uniformly concluded. Many heterocycles, especially amines, were identified early on, and received trivial names which are still preferred. Some monocyclic compounds of this kind are shown in the following chart, with the common (trivial) name in bold and a systematic name based on the Hantzsch-Widman system given beneath it in blue. The rules for using this system will be given later. For most students, learning these common names will provide an adequate nomenclature background.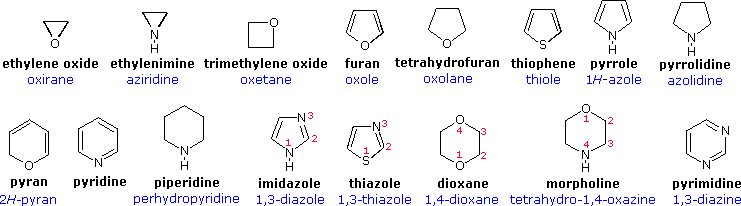
| Element | oxygen | sulfur | selenium | nitrogen | phosphorous | silicon | boron |
|---|---|---|---|---|---|---|---|
| Valence | II | II | II | III | III | IV | III |
| Prefix | Oxa | Thia | Selena | Aza | Phospha | Sila | Bora |
| Ring Size | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|
| Suffix Unsaturated Saturated | irene irane | ete etane | ole olane | ine inane | epine epane | ocine ocane | onine onane | ecine ecane |
• Saturated 3, 4 & 5-membered nitrogen heterocycles should use respectively the traditional "iridine", "etidine" & "olidine" suffix.
• Unsaturated nitrogen 3-membered heterocycles may use the traditional "irine" suffix.
• Consistent use of "etine" and "oline" as a suffix for 4 & 5-membered unsaturated heterocycles is prevented by their former use for similar sized nitrogen heterocycles.
• Established use of oxine, azine and silane for other compounds or functions prohibits their use for pyran, pyridine and silacyclohexane respectively.
Preparation and Reactions
Three-Membered Rings
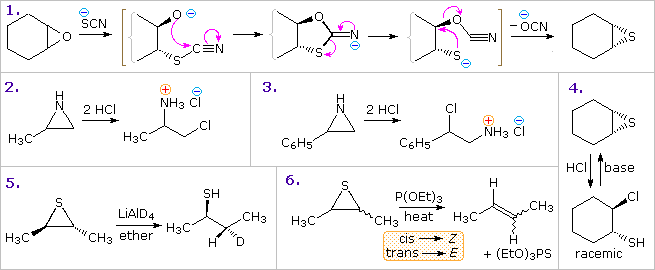
Oxiranes (epoxides) are the most commonly encountered three-membered heterocycles. Epoxides are easily prepared by reaction of alkenes with peracids, usually with good stereospecificity. Because of the high angle strain of the three-membered ring, epoxides are more reactive that unstrained ethers. Addition reactions proceeding by electrophilic or nucleophilic opening of the ring constitute the most general reaction class. Example 1 in the following diagram shows one such transformation, which is interesting due to subsequent conversion of the addition intermediate into the corresponding thiirane. The initial ring opening is stereoelectronically directed in a trans-diaxial fashion, the intermediate relaxing to the diequatorial conformer before cyclizing to a 1,3-oxathiolane intermediate.Other examples show similar addition reactions to thiiranes and aziridines. The acid-catalyzed additions in examples 2 and 3, illustrate the influence of substituents on the regioselectivity of addition. Example 2 reflects the SN2 character of nucleophile (chloride anion) attack on the protonated aziridine (the less substituted carbon is the site of addition). The phenyl substituent in example 3 serves to stabilize the developing carbocation to such a degree that SN1 selectivity is realized. The reduction of thiiranes to alkenes by reaction with phosphite esters (example 6) is highly stereospecific, and is believed to take place by an initial bonding of phosphorous to sulfur.
An interesting regioselectivity in the intramolecular ring-opening reactions of disubstituted epoxides having a pendant γ-hydroxy substituent has been noted. As illustrated below, acid and base-catalyzed reactions normally proceed by 5-exo-substitution (reaction 1), yielding a tetrahydrofuran product. However, if the oxirane has an unsaturated substituent (vinyl or phenyl), the acid-catalyzed opening occurs at the allylic (or benzylic) carbon (reaction 2) in a 6-endo fashion. The π-electron system of the substituent assists development of positive charge at the adjacent oxirane carbon, directing nucleophilic attack to that site.
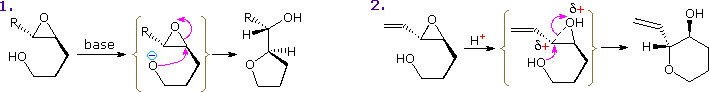
Four-Membered Rings
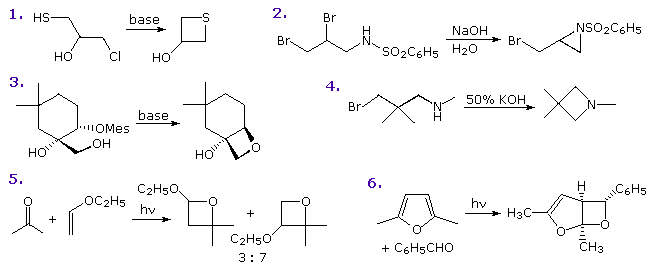
PreparationSeveral methods of preparing four-membered heterocyclic compounds are shown in the following diagram. The simple procedure of treating a 3-halo alcohol, thiol or amine with base is generally effective, but the yields are often mediocre. Dimerization and elimination are common side reactions, and other functions may compete in the reaction. In the case of example 1, cyclization to an oxirane competes with thietane formation, but the greater nucleophilicity of sulfur dominates, especially if a weak base is used. In example 2 both aziridine and azetidine formation are possible, but only the former is observed. This is a good example of the kinetic advantage of three-membered ring formation. Example 4 demonstrates that this approach to azetidine formation works well in the absence of competition. Indeed, the exceptional yield of this product is attributed to the gem-dimethyl substitution, the Thorpe-Ingold effect, which is believed to favor coiled chain conformations. The relatively rigid configuration of the substrate in example 3, favors oxetane formation and prevents an oxirane cyclization from occurring. Finally, the Paterno-Buchi photocyclizations in examples 5 and 6 are particularly suited to oxetane formation.
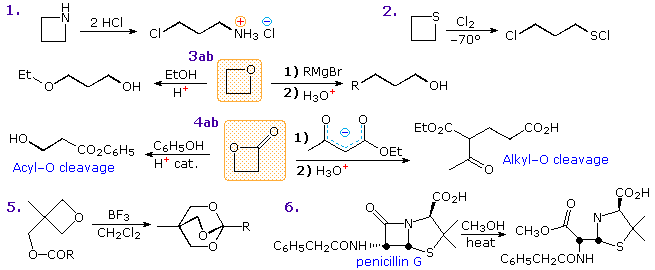
Reactions of four-membered heterocycles also show the influence of ring strain. Some examples are given in the following diagram. Acid-catalysis is a common feature of many ring-opening reactions, as shown by examples 1, 2 & 3a. In the thietane reaction (2), the sulfur undergoes electrophilic chlorination to form a chlorosulfonium intermediate followed by a ring-opening chloride ion substitution. Strong nucleophiles will also open the strained ether, as shown by reaction 3b. Cleavage reactions of β-lactones may take place either by acid-catalyzed acyl exchange, as in 4a, or by alkyl-O rupture by nucleophiles, as in 4b. Example 5 is an interesting case of intramolecular rearrangement to an ortho-ester. Finally, the β-lactam cleavage of penicillin G (reaction 6) testifies to the enhanced acylating reactivity of this fused ring system. Most amides are extremely unreactive acylation reagents, thanks to stabilization by p-π resonance. Such electron pair delocalization is diminished in the penicillins, leaving the nitrogen with a pyramidal configuration and the carbonyl function more reactive toward nucleophiles.
Five-Membered Rings
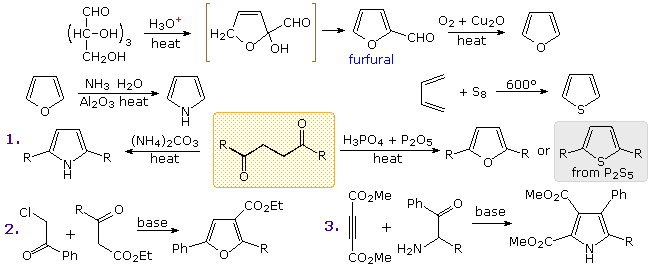
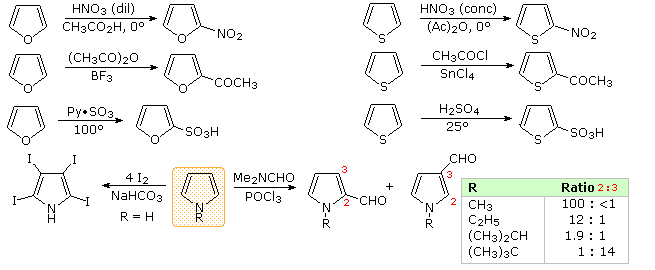
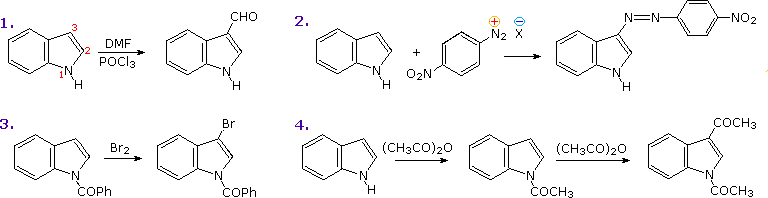
PreparationCommercial preparation of furan proceeds by way of the aldehyde, furfural, which in turn is generated from pentose containing raw materials like corncobs, as shown in the uppermost equation below. Similar preparations of pyrrole and thiophene are depicted in the second row equations. Equation 1 in the third row illustrates a general preparation of substituted furans, pyrroles and thiophenes from 1,4-dicarbonyl compounds, known as the Paal-Knorr synthesis. Many other procedures leading to substituted heterocycles of this kind have been devised. Two of these are shown in reactions 2 and 3. Furan is reduced to tetrahydrofuran by palladium-catalyzed hydrogenation. This cyclic ether is not only a valuable solvent, but it is readily converted to 1,4-dihalobutanes or 4-haloalkylsulfonates, which may be used to prepare pyrrolidine and thiolane.
Dipolar cycloaddition reactions often lead to more complex five-membered heterocycles.
Reactions
The chemical reactivity of the saturated members of this class of heterocycles: tetrahydrofuran, thiolane and pyrrolidine, resemble that of acyclic ethers, sulfides, and 2º-amines, and will not be described here. 1,3-Dioxolanes and dithiolanes are cyclic acetals and thioacetals. These units are commonly used as protective groups for aldehydes and ketones, and may be hydrolyzed by the action of aqueous acid.
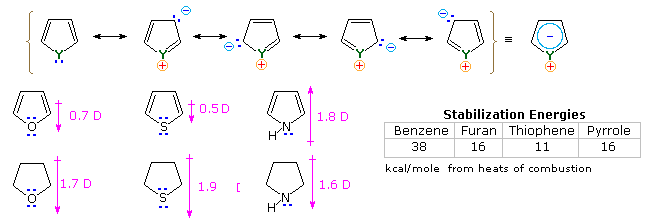
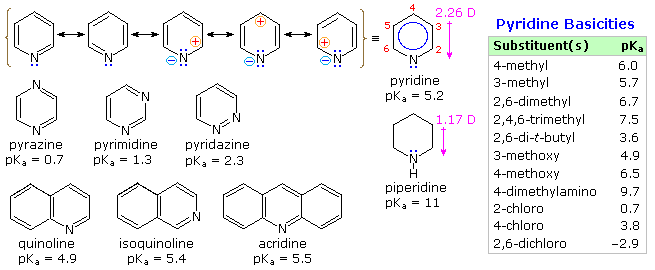
It is the "aromatic" unsaturated compounds, furan, thiophene and pyrrole that require our attention. In each case the heteroatom has at least one pair of non-bonding electrons that may combine with the four π-electrons of the double bonds to produce an annulene having an aromatic sextet of electrons. This is illustrated by the resonance description at the top of the following diagram. The heteroatom Y becomes sp2-hybridized and acquires a positive charge as its electron pair is delocalized around the ring. An easily observed consequence of this delocalization is a change in dipole moment compared with the analogous saturated heterocycles, which all have strong dipoles with the heteroatom at the negative end. As expected, the aromatic heterocycles have much smaller dipole moments, or in the case of pyrrole a large dipole in the opposite direction. An important characteristic of aromaticity is enhanced thermodynamic stability, and this is usually demonstrated by relative heats of hydrogenation or heats of combustion measurements. By this standard, the three aromatic heterocycles under examination are stabilized, but to a lesser degree than benzene.
Additional evidence for the aromatic character of pyrrole is found in its exceptionally weak basicity (pKa ca. 0) and strong acidity (pKa = 15) for a 2º-amine. The corresponding values for the saturated amine pyrrolidine are: basicity 11.2 and acidity 32.
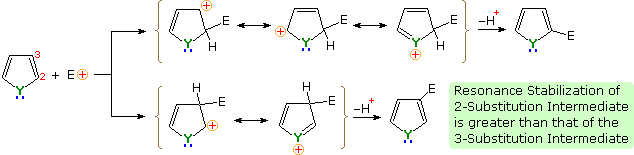
An explanation for the general α-selectivity of these substitution reactions is apparent from the mechanism outlined below. The intermediate formed by electrophile attack at C-2 is stabilized by charge delocalization to a greater degree than the intermediate from C-3 attack. From the Hammond postulate we may then infer that the activation energy for substitution at the former position is less than the latter substitution.
The bottom line of the new diagram illustrates the remarkable influence that additional nitrogen units have on the hydrolysis of a series of N-acetylazoles in water at 25 ºC and pH=7. The pyrrole compound on the left is essentially unreactive, as expected for an amide, but additional nitrogens markedly increase the rate of hydrolysis. This effect has been put to practical use in applications of the acylation reagent 1,1'-carbonyldiimidazole (Staab's reagent).
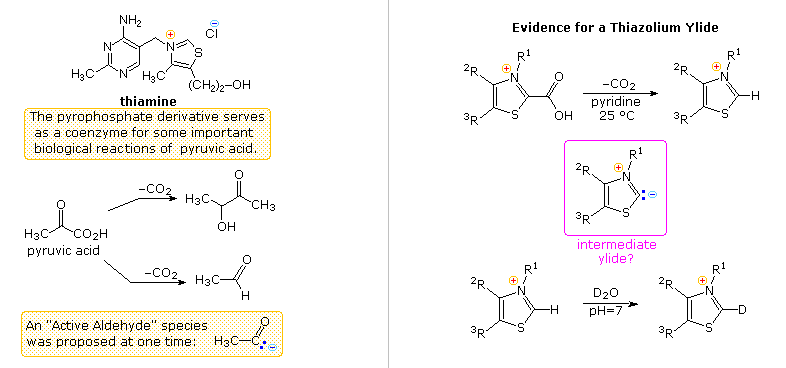
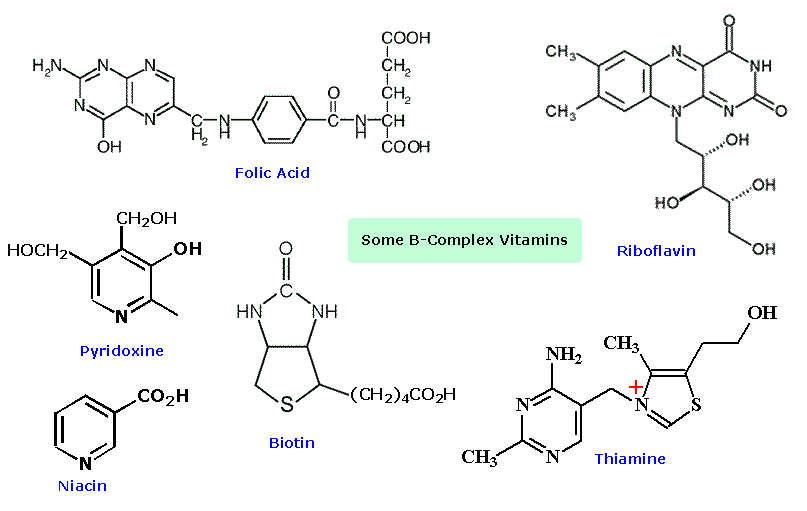
Another facet of heterocyclic chemistry was disclosed in the course of investigations concerning the action of thiamine (following diagram). As its pyrophosphate derivative, thiamine is a coenzyme for several biochemical reactions, notably decarboxylations of pyruvic acid to acetaldehyde and acetoin. Early workers speculated that an "active aldehyde" or acyl carbanion species was an intermediate in these reactions. Many proposals were made, some involving the aminopyrimidine moiety, and others, ring-opened hydrolysis derivatives of the thiazole ring, but none were satisfactory. This puzzle was solved when R. Breslow (Columbia) found that the C-2 hydrogen of thiazolium salts was unexpectedly acidic (pKa ca. 13), forming a relatively stable ylide conjugate base. As shown, this rationalizes the facile decarboxylation of thiazolium-2-carboxylic acids and deuterium exchange at C-2 in neutral heavy water.
Appropriate thiazolium salts catalyze the conversion of aldehydes to acyloins in much the same way that cyanide ion catalyzes the formation of benzoin from benzaldehyde, the benzoin condensation. By clicking on the diagram, a new display will show mechanisms for these two reactions. Note that in both cases an acyl anion equivalent is formed and then adds to a carbonyl function in the expected manner. The benzoin condensation is limited to aromatic aldehydes, but the use of thiazolium catalysts has proven broadly effective for aliphatic and aromatic aldehydes. This approach to acyloins employs milder conditions than the reduction of esters to enediol intermediates by the action of metallic sodium .
Six-Membered Rings
PropertiesThe chemical reactivity of the saturated members of this class of heterocycles: tetrahydropyran, thiane and piperidine, resemble that of acyclic ethers, sulfides, and 2º-amines, and will not be described here. 1,3-Dioxanes and dithianes are cyclic acetals and thioacetals. These units are commonly used as protective groups for aldehydes and ketones, as well as synthetic intermediates, and may be hydrolyzed by the action of aqueous acid. The reactivity of partially unsaturated compounds depends on the relationship of the double bond and the heteroatom (e.g. 3,4-dihydro-2H-pyran is an enol ether).
Fully unsaturated six-membered nitrogen heterocycles, such as pyridine, pyrazine, pyrimidine and pyridazine, have stable aromatic rings. Oxygen and sulfur analogs are necessarily positively charged, as in the case of 2,4,6-triphenylpyrylium tetrafluoroborate.
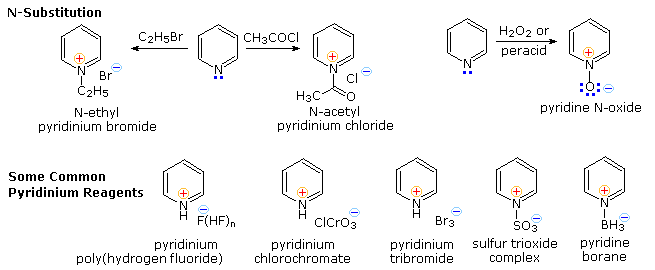
Electrophilic Substitution of Pyridine
Pyridine is a modest base (pKa=5.2). Since the basic unshared electron pair is not part of the aromatic sextet, as in pyrrole, pyridinium species produced by N-substitution retain the aromaticity of pyridine. As shown below, N-alkylation and N-acylation products may be prepared as stable crystalline solids in the absence of water or other reactive nucleophiles. The N-acyl salts may serve as acyl transfer agents for the preparation of esters and amides. Because of the stability of the pyridinium cation, it has been used as a moderating component in complexes with a number of reactive inorganic compounds. Several examples of these stable and easily handled reagents are shown at the bottom of the diagram. The poly(hydrogen fluoride) salt is a convenient source of HF for addition to alkenes and conversion of alcohols to alkyl fluorides, pyridinium chlorochromate (PCC) and its related dichromate analog are versatile oxidation agents and the tribromide salt is a convenient source of bromine. Similarly, the reactive compounds sulfur trioxide and diborane are conveniently and safely handled as pyridine complexes.
Amine oxide derivatives of 3º-amines and pyridine are readily prepared by oxidation with peracids or peroxides, as shown by the upper right equation. Reduction back to the amine can usually be achieved by treatment with zinc (or other reactive metals) in dilute acid.
Pyridine N-oxide undergoes some electrophilic substitutions at C-4 and others at C-3. The coordinate covalent N–O bond may exert a push-pull influence, as illustrated by the two examples on the right. Although the positively charged nitrogen alone would have a strong deactivating influence, the negatively charged oxygen can introduce electron density at C-2, C-4 & C-6 by π-bonding to the ring nitrogen. This is a controlling factor in the relatively facile nitration at C-4. However, if the oxygen is bonded to an electrophile such as SO3, the resulting pyridinium ion will react sluggishly and preferentially at C-3.
The fused ring heterocycles quinoline and isoquinoline provide additional evidence for the stability of the pyridine ring. Vigorous permanganate oxidation of quinoline results in predominant attack on the benzene ring; isoquinoline yields products from cleavage of both rings. Note that naphthalene is oxidized to phthalic acid in a similar manner. By contrast, the heterocyclic ring in both compounds undergoes preferential catalytic hydrogenation to yield tetrahydroproducts. Electrophilic nitration, halogenation and sulfonation generally take place at C-5 and C-8 of the benzene ring, in agreement with the preceding description of similar pyridine reactions and the kinetically favored substitution of naphthalene at C-1 (α) rather than C-2 (β).
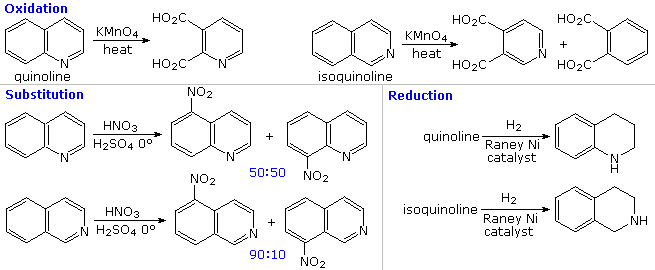
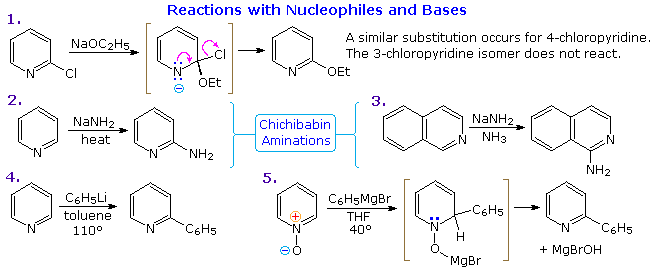
Thanks to the nitrogen in the ring, pyridine compounds undergo nucleophilic substitution reactions more easily than equivalent benzene derivatives. In the following diagram, reaction 1 illustrates displacement of a 2-chloro substituent by ethoxide anion. The addition-elimination mechanism shown for this reaction is helped by nitrogen's ability to support a negative charge. A similar intermediate may be written for substitution of a 4-halopyridine, but substitution at the 3-position is prohibited by the the failure to create an intermediate of this kind. The two Chichibabin aminations in reactions 2 and 3 are remarkable in that the leaving anion is hydride (or an equivalent). Hydrogen is often evolved in the course of these reactions. In accord with this mechanism, quinoline is aminated at both C-2 and C-4.
Addition of strong nucleophiles to N-oxide derivatives of pyridine proceed more rapidly than to pyridine itself, as demonstrated by reactions 4 and 5. The dihydro-pyridine intermediate easily loses water or its equivalent by elimination of the –OM substituent on nitrogen.
Some Polycyclic Heterocycles
Heterocyclic structures are found in many natural products. Examples of some nitrogen compounds, known as alkaloids because of their basic properties, were given in the amine chapter. Some other examples are displayed in the following diagram. Camptothecin is a quinoline alkaloid which inhibits the DNA enzyme topoisomerase I. Reserpine is an indole alkaloid, which has been used for the control of high blood pressure and the treatment of psychotic behavior. Ajmaline and strychnine are also indole alkaloids, the former being an antiarrhythmic agent and latter an extremely toxic pesticide. The neurotoxins saxitoxin and tetrodotoxin both have marine origins and are characterized by guanidiniun moieties. Aflatoxin B1 is a non-nitrogenous carcinogenic compound produced by the Aspergillus fungus.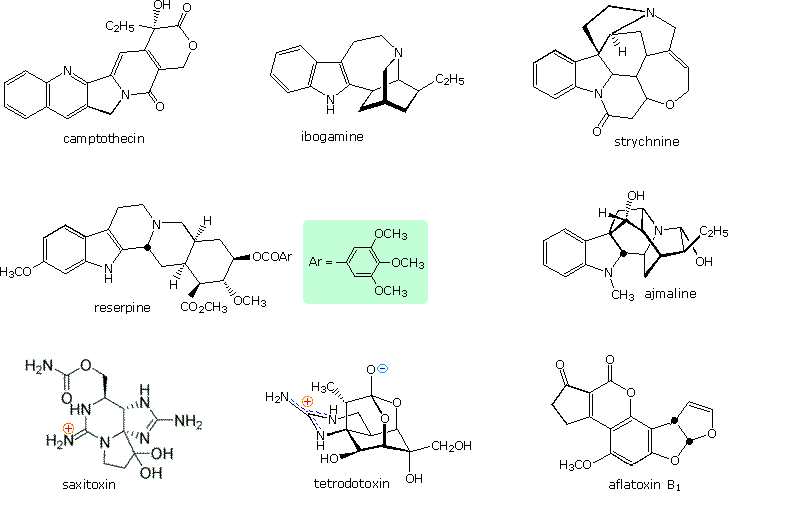
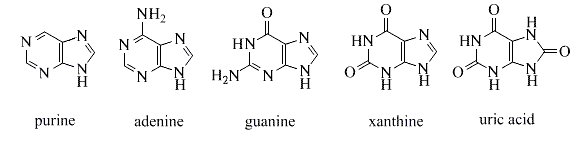
Derivatives of the simple fused ring heterocycle purine constitute an especially important and abundant family of natural products. The amino compounds adenine and guanine are two of the complementary bases that are essential components of DNA. Structures for these compounds are shown in the following diagram. Xanthine and uric acid are products of the metabolic oxidation of purines. Uric acid is normally excreted in the urine; an excess serum accumulation of uric acid may lead to an arthritic condition known as gout.





As a global Contract Research Organization (CRO), headquartered in New York, USA, Alfa Chemistry has served the pharmaceutical and biotechnology industries for eight years. copal gum
ReplyDeleteThanks for sharing the crucial article to me, It would be helpful for me.
ReplyDeleteRegards,
upcoming schools in Aurangabad | bhondawe patil public school